 Journal for Advances in Science
Journal for Advances in Science
Research article | Published November 28, 2022 | Original research published in Nature Neuroscience
Catching the Epigenome in Action: A Global View on Gene Regulatory Logic in the Mouse Cortex
Christina Lilliehook
Christina Lilliehook is a Senior Science Writer at RA Capital, a Science Editor at Life Science Editors, and a former Deputy Editor at Cell Stem Cell.
Original research: Noack, F., Vangelisti, S., Raffl, G. et al. Multimodal profiling of the transcriptional regulatory landscape of the developing mouse cortex identifies Neurog2 as a key epigenome remodeler. Nat Neurosci 25, 154–167 (2022). https://doi.org/10.1038/s41593-021-01002-4
Subjects: Developmental neurogenesis, Epigenomics, Gene regulation, Gene regulatory networks
New Ground article reviewed by: Boyan Bonev
How dividing cells acquire different somatic cell fates in the early embryo continues to fascinate scientists. This process begins after fertilization, i.e., when the male and female genomes have combined and the egg has transformed into a sphere of rapidly dividing cells. In developing organisms, cells balance proliferation and differentiation into specific somatic cell fates. Although cellular properties likely change only gradually, scientists extrapolate cellular states on the basis of proliferation capacity, molecular profiles, and morphology to create cell state trajectories of linear differentiation. Generally, tissue-specific stem cells are considered to transition from a stem cell state to one or more progenitor states, and ultimately to a non-proliferative cell state with mature functional properties.
While gaining a precise molecular understanding of the earliest cell fate decisions (when stem cells are few) will likely remain out of reach, developmental scientists have defined several transcription factors (TFs) that drive the formation of specific cell types and tissues, referred to as lineage-specific TFs. For instance, Tal1 is known to drive blood and endothelial cell fate, whereas Nk2-5, Gata4 and Tbx5 influence the formation of cardiac tissue. In the nervous system, Sox2 and Neurog2 (neurogenin 2), among others, play an important role in the acquisition of neuronal cell fate.
Mapping the gene regulatory landscape of neural differentiation in the neocortex
Gene regulatory networks (GRNs) are at the very core of cell fate decisions. Accordingly, GRNs are responsible for starting, stopping and fine-tuning all cellular processes in an organism. They are epigenomic features, i.e., they leave the sequence of the DNA unchanged, but govern – through interactions between the DNA, RNA and proteins – the expression levels of target genes in time and space. It’s also important to bear in mind that GRNs are not directly detectable, but for the most part inferred by statistical analysis of gene expression data. Wet lab experiments are therefore needed to validate causal relationships.
In a recent Nature Neuroscience publication, “Multimodal profiling of the transcriptional regulatory landscape of the developing mouse cortex identifies Neurog2 as a key epigenome remodeler,” Boyan Bonev and his team map the gene regulatory landscape of neural differentiation in the mouse neocortex and identify new GRN elements and how they influence neuronal lineage acquisition within the three-dimensional genome structure. They also validate their predictions in the functional in vivo setting.
So how does gene regulation work? In prokaryotes, gene regulation is generally simple: TFs bind to short promoter regions on the DNA that define where transcription of a gene begins. In eukaryotes, and especially mammals, promoter regions are also close to the gene they regulate, but gene regulation is highly complex: initiation of transcription can occur when TFs bind to promoters that are physically accessible, or “open”; but transcription is also tuned to match cellular needs by regulatory proteins and short regions of DNA called enhancers, or cis-regulatory elements, that bind to TFs to form large regulatory protein complexes. Frequently, individual genes are even regulated by multiple enhancers.
The vast cellular diversity required to create larger and more complex species is believed to be based on the evolution of enhancers as part of the regulatory control of transcription. Indeed, enhancers are considered the key building blocks of GRNs. Often distal to the gene they regulate, enhancer regions distort the shape of the chromatin (linear DNA wrapped around histones) to form what are known as chromatin loops. Such loops enable them to physically interact with promoters close to the gene they regulate.
Limitations of single-cell technologies
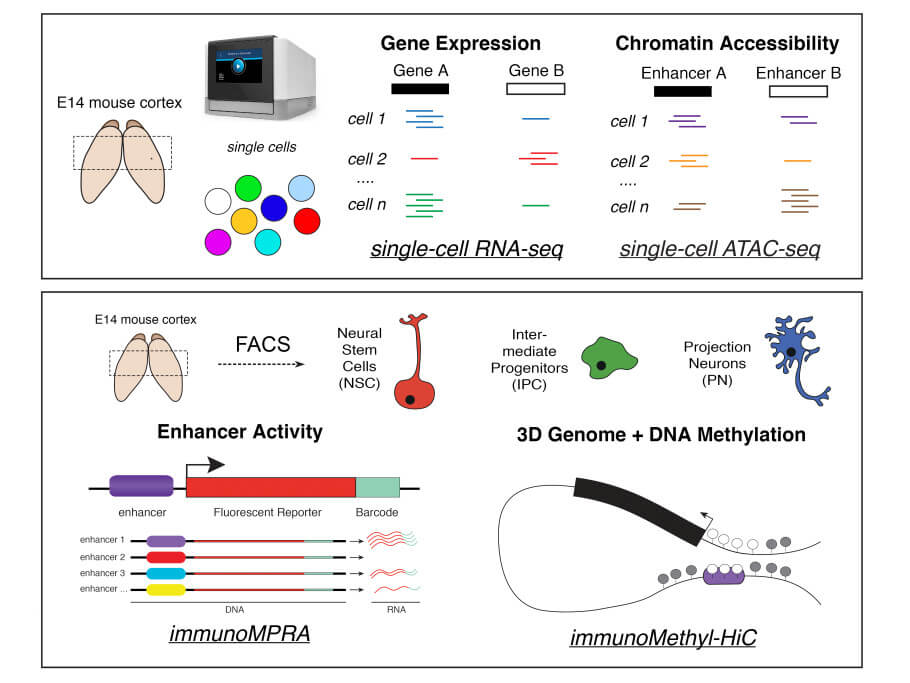
In recent years, single-cell technologies have revolutionized the study of GRNs, allowing scientists to decipher how the epigenome changes in dividing cells in a way that permits and shapes lineage acquisition. Several groups have created transcriptomic landscapes of the developing and adult mouse and human brain by employing scRNA-seq (single-cell RNA sequencing) and scATAC-seq (single-cell transposase-accessible chromatin sequencing) analyses, the latter being used to identify chromatin regions accessible to binding by regulatory elements.
scRNA-seq and scATAC-seq technologies, however, both have their limitations. Neither can measure dynamic changes over time; they can only capture snapshots by gathering information from individual cells at different stages of development. Subsequently, researchers rely on computational approaches to predict the temporal sequence of developmental stages. Other relevant modalities of epigenetic regulation, however, such as changes in DNA methylation and physical proximity, cannot be readily extrapolated from such data. As such, scRNA-seq and scATAC-seq studies alone cannot definitively answer the question of how enhancer regions engage with TFs in the dynamic three-dimensional DNA environment to regulate transcription and determine cell fate.
Single-cell catalog of novel enhancer-gene pairs
To overcome these limitations, the Bonev Lab recently employed a multimodal epigenomics approach in an effort to find out exactly how GRNs in the mouse embryonic cortex are formed and remodeled to allow neural stem cells to differentiate and become mature neurons. The team first reconstructed the temporal sequence of neural cell development, i.e., its lineage trajectory, on the basis of the cells’ respective transcriptomes detected by scRNA-seq analysis, and, separately, on the basis of the accessibility of known marker genes in the scATAC-seq data. At this stage of the analysis, they were able to identify which TFs become bound to accessible chromatin in specific cell states throughout cortical development. They also determined that the accessibility of the chromatin between cell states changed the most in enhancer regions, indicating that enhancers are more cell-type-specific than are promoters.
To identify enhancer-gene pairs, the authors computationally integrated scRNA and scATAC data to search for correlations between changes in enhancer accessibility and changes in gene expression of the enhancer’s predicted target. Thus, they were able to assemble a rich catalog of novel enhancer-gene pairs in the developing cortex that were specific to different developmental states of cells: to neural stem cells, to intermediate progenitor cells (which usually divide once more to produce two neurons, thus expanding the progenitor pool), or to mature projection neurons that are terminally differentiated.
The authors found that TFs known to be specific for neural stem cells, such as Sox2, Tead and AP-1 (Fos/Jun), preferentially became bound to enhancers in neural stem cells but were largely absent from enhancers in mature neurons. Conversely, they found that neurogenic TFs such as Neurog2 and Eomes were enriched in enhancers of intermediate progenitor cells and depleted in those of neural stem cells. These findings confirm that enhancers forming GRNs with TFs known to drive the acquisition of neural stem cell fate are only accessible in neural stem cells. And conversely, that enhancers forming GRNs with TFs that drive mature neuronal fate are accessible in mature neurons, but not in neural stem cells. At a broader level, the single-cell catalog of novel enhancer-gene pairs opened the door for the authors to investigate how the inner workings of gene regulatory networks change depending on the respective cell state and how they are influenced by the newly identified enhancers.
Functional validation of measurements of chromatin accessibility
However, a major limitation of the existing single-cell epigenomic studies is that such predictions regarding enhancer activity and specificity based on biochemical measurements of chromatin accessibility are rarely functionally validated or, when they are, only a few examples are selected. To address this important shortcoming, the authors employed a massively parallel reporter assay (MPRA), which allows direct measurement of enhancer activity at very high throughput. Importantly, to date MPRAs have primarily been used in vitro, while here the authors were able to directly evaluate enhancer activity in vivo by combining MPRA with in utero electroporation, a non-invasive gene transfer method commonly used to study brain development in mice. Furthermore, they were able to assess enhancer specificity by isolating the three major cell populations in the developing cortex (neural stem cells, intermediate progenitors, and mature neurons), and to identify their respective catalogs of enhancers, the vast majority of which were previously unknown.
When they zoomed in on specific genomic binding motifs in specific cell states, they found that active enhancers matched with TF expression, whereas inactive enhancers matched with TF repression. Importantly, to directly assess whether TF binding is causal for enhancer activity, the authors mutated the binding sites for several TFs and found that for some, but not all, doing so completely eliminated enhancer activity. For example, when the Neurog2 binding motif was mutated in an enhancer region specifically active in intermediate progenitor cells (and one that normally drives the transcription of the Delta-like 1 gene, which is linked with human neurodevelopmental disorders, see here), transcription was eliminated. These findings underscore that expanding our knowledge of how cell-type-specific enhancers are regulated can help us understand not only basic biology, but also the mechanisms underlying disease pathology.
Enhancer-promoter interactions beyond the basic model
Many studies have shown that enhancer-promoter interactions are generally correlated with gene expression, and also, that such interactions are highly dynamic. However, there is still no easy way to quantify enhancer-promoter interactions in living organs and tissues. Gene transcription, therefore, has been assumed to activate when stable enhancer-promoter loops are formed. Recently, a few reports have challenged this simplistic activity-by-contact model. In fact, some enhancers have been reported to form weak or no interactions at all with their promoter. For instance (see here), researchers identified a chromosome conformation in embryonic stem cells that prevented physical interaction between a specific enhancer and its promoter, yet this interaction was required to drive transcription of the Sonic hedgehog gene. Obviously, there must be another process in place to allow transcription. Similarly, when investigating dorsoventral patterning in the fly embryo (a developmental process in which the belly and back regions of the embryo become distinct from one another), other researchers found (see here) that spatial chromatin distribution did not differ between tissues, despite varying expression patterns across tissues as detected by scRNA-seq. In this case, again, the interaction between enhancer and promoter is not necessarily determined by physical proximity.
To learn more about the kinetics and strength of enhancer-promoter interactions beyond the basic model, Boyan Bonev and his team isolated state-specific populations of neural stem cells, intermediate progenitor cells and projection neurons from the embryonic mouse cortex and conducted a Methyl-HiC analysis. This technique jointly analyzes DNA methylation and the spatial organization of chromosomes (the methylation of the DNA sequence of a gene may turn the gene off, while demethylation may turn it on). Hence, the authors were able to analyze the methylation status of the enhancer-gene pairs they identified based on their integrated single-cell analysis, and to measure the interaction frequency (or physical proximity within the nucleus) between these elements in different cell types.
The authors observed that promoters remained consistently demethylated, regardless of whether the associated gene was upregulated or repressed during neural differentiation. Instead, it was at the enhancers that DNA methylation levels were found to be dynamic and correlated with changes in accessibility. When examining the physical proximity between enhancers and promoters, the authors observed that their interactions were generally dynamic and stronger in cell types characterized by an active enhancer and an expressed gene, thus supporting the textbook model.
However, they also observed that many enhancer-promoter pairs deviated from this model and were either looped before transcription occurred or became even more distal upon gene activation. Importantly, these observations can reconcile the contradictory findings on the importance of enhancers’ physical proximity for gene activation: although the classical model appears to be true genome-wide (at least in the system studied), there are many exceptions which could explain the results seen at individual loci, e.g. the Sonic hedgehog locus. They were unable to predict either DNA methylation levels or enhancer-promoter interactions solely from scRNA-seq and scATAC-seq, further demonstrating the importance of profiling additional epigenetic modalities to study GRNs.
To better understand how such regulatory interactions were remodeled throughout neuronal differentiation, the authors quantified the physical interaction strength and specificity for each TF. Unexpectedly, they found that several TFs that were cell-state-specific were also associated with strong looping. Of these, they chose to focus on Neurog2, as it was correlated with both chromatin interaction and DNA methylation, and given its relationship with the Eomes locus, which encodes for Eomesodermin/Tbr2, a TF expressed in intermediate progenitor cells.
To investigate whether Neurog2 can functionally induce epigenome changes, Bonev’s team went on to overexpress Neurog2 in the embryonic mouse cortex. Consistent with earlier work and the computational analysis, Neurog2 overexpression resulted in both Neurog2 binding sites and motifs becoming more accessible. In addition, the team found that sites that bound Neurog2 also had stronger interactions with each other but that the overall spatial organization of the genome did not change, suggesting that these effects are specific to Neurog2. Gene expression was increased when Neurog2 was bound to either the promoter or the enhancer region but synergistically increased when it was bound to both. For the Eomes locus specifically, contacts between enhancer and promoter regions increased only when Neurog2 was bound to the enhancers. Taken together, the overexpression experiments suggest that the synergistic activation induced by binding to both the promoter and the enhancer, and the formation of a chromatin loop, can lead to stronger gene activation and potentially a more robust cell fate switch.
Conclusion
This work yields several important insights. Firstly, the study shows that despite the richness of the enhancer landscape in the developing cortex, only a few enhancers actually drive neuronal lineage priming. The authors conclude that such pioneering activity is highly context-dependent and shaped by specific enhancer properties and cofactors – similar to the pioneering activity of Foxa2 in the endoderm.
Secondly, the study is the first to combine DNA methylation analysis and epigenome architecture analysis with scRNA-seq and scATAC-seq. In addition to revealing that the interaction strength between enhancers and promoters varies in a way that can’t necessarily be predicted based on chromatin accessibility or gene expression, they find that interactions between enhancers and promoters are largely cell-state-specific and quantitative, as opposed to strictly on or off.
Thirdly, the authors propose that a subset of TFs that are associated with dynamic chromatin looping, as exemplified by Neurog2, are capable of coordinating epigenome remodeling and thereby shaping the inner workings of GRNs. Notably, they conclude – in agreement with a 2021 publication in Nature (see here) – that the earliest neural cell fate transitions (from neural stem cells to intermediate progenitors and then to postnatal neurons) are largely preserved and that neural differentiation programs primarily diverge in mature, postmitotic neurons.
Lastly, the study highlights the importance of conducting multimodal epigenome studies to catch the dynamic epigenome in action.
How to reuse
The CC BY 4.0 license requires re-users to give due credit to the creator. It allows re-users to distribute, remix, adapt, and build upon the material in any medium or format, even for commercial purposes.
You can reuse an article (e.g. by copying it to your news site) by adding the following line:
Catching the Epigenome in Action: A Global View on Gene Regulatory Logic in the Mouse Cortex © 2022 by Christina Lilliehook is licensed under Attribution 4.0 International
Or by simply adding:
Article
© 2022 by Christina Lilliehook / CC BY
To learn more about the available options, and for details, please consult New Ground’s How to reuse section.

This article – but not the graphics or images – is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
This article – but not the graphics or images – is licensed under a Creative Commons Attribution 4.0 License.